Decision-Based Evidence Making
Developing tools and strategies for comparative effectiveness

Despite the publication of over 18,000 Randomised Clinical Trials (RCTs) each year, available clinical evidence is often of limited quality. Generating the evidence needed to support an evidence-based healthcare system will require collective effort, and needs to be driven by decision makers in the healthcare community such as patients, physicians, policymakers and payers.
Despite the steady increase in public and private funding of clinical trials and health services, the current research enterprise in the US is not meeting the rising demand from decision makers for studies demonstrating evidence of clinical effectiveness. According to the National Library of Medicine, results from over 18,000 randomised clinical trials were published in 2007 along with numerous other non-experimental studies, all intended to provide information about ‘what works in healthcare.’ Despite this healthy output of scientific literature, the majority of systematic literature reviews, technology assessments and clinical practice guidelines that evaluate all available published literature on virtually any topic have concluded that the available evidence is ‘limited’ and that many published studies are of ‘low quality.’ For these reasons, these reviews generally conclude that the available evidence does not support reliable conclusions about the most important clinical and policy questions related to the topic reviewed. This paradox—the large volume of clinical and health services research and the low quality of evidence—requires some explanation if one is hoping to move further toward evidence-based clinical and health policy decision making. It is, therefore, important to understand the nature of the gaps in evidence; how and why does the current body of research predictably fall short? This is particularly important as the US Federal government has recently decided to invest billions of additional dollars in clinical and health services research, including US$ 1.1 billion for comparative effectiveness research.
There are some key observations that provide insight into the apparent discrepancy between the volume and the quality of evidence. Systematic reviews, clinical guidelines and health technology assessments review evidence with the goal of informing decision makers—primarily patients, clinicians, payers, and policymakers. The process of conducting a review begins with a deliberate effort to identify the critical questions faced by these decision makers, and the literature search and appraisal process is conducted with reference to these key questions. Viewed through this prism, they commonly conclude that the available evidence on these key questions is limited. The process of generating evidence through clinical and health services research does not usually begin with a careful assessment of what information decision makers most need to know. Most research is investigator initiated, with topics selected and studies designed with reference to previously published studies and through dialogue with other researchers working on related issues. Decision makers are rarely involved in the process of refining study questions or the actual design of the studies. In those infrequent cases where they are included in some of these early discussions, their unfamiliarity with the technical content of research may limit their ability to participate meaningfully in the discussion. Given these very different starting points and perspectives, it is unsurprising that systematic literature reviews, which are guided by the questions faced by decision makers, generally highlight that these questions are not consistently addressed by research because it was designed and implemented without attention to or involvement of these decision makers.
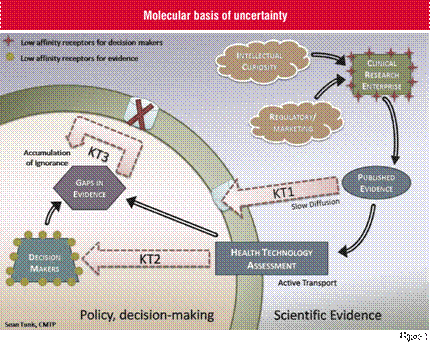
The molecular basis of uncertainty
An analogy from molecular biology may be useful in communicating the disconnect that exists between those who generate scientific evidence and those who utilise this evidence to make clinical and health policy decisions. Figure 1 depicts the cycle of information flow from the ‘extracellular environment’ of the clinical research enterprise to the ‘intracellular world’ that is inhabited by the decision makers. On one side of the cell membrane is the realm of those who generate new scientific evidence through clinical and health services research—guided for the most part by their intellectual curiosity, and not particularly attentive to the needs of those on the other side of the cell wall. The evidence produced by these researchers encounters a number of barriers during the process of ‘diffusion’ across the membrane and into the cell. The first barrier in Knowledge Transfer (KT1) involves the commonly observed slow translation of knowledge into practice and policy, resulting in a lengthy time-lag between the publication of new evidence and the impact of that evidence on what is actually done. The speed of translation can sometimes be increased when scientific evidence is compiled and analysed through Health Technology Assessment (HTA) organisations (KT2), and this active transport mechanism is an important pathway by which the linkage between evidence and decision making can be considerably enhanced. Once decision makers have applied the available evidence to their decisions, they will frequently observe the gaps in knowledge on critical questions. Ideally, these unanswered questions, or areas of ignorance, would be fed back to the research community so that further research could be focussed on these issues. However, a defective transport mechanism (KT3) severely impairs the communication of these research priorities to the clinical research enterprise. This defective transport mechanism ensures that many of these important questions remain unanswered, leading to an accumulation of ignorance which surrounds the decision makers. Potential interventions to improve the cycle of information flow between the intra and extra-cellular space will need to focus in part on improving the consistency with which the unanswered questions of decision makers are effectively communicated to the clinical and health services research community, and become a higher priority for attention.
Tools and strategies for decision-based evidence making
The molecular model suggests that there is a need for tools and strategies through which the link between decision makers and researchers can be strengthened. While there have been numerous informal and ad hoc efforts in this direction to increase dialogue between decision makers and researchers, such informal interactions are inconsistent in their ability to produce relevant and timely information for decision making. One general requirement is that communication about important gaps in evidence and the appropriate design of research to address those gaps must take place long before there is a decision to be made. When payers and policy makers are faced with a specific decision, it is generally much too late to begin a conversation about what sort of evidence would be useful. A number of tools and strategies intended to support ‘decision-based evidence making’ are currently being piloted the US-based Center for Medical Technology Policy (CMTP), a private, non-profit organisation that provides a neutral forum in which patients, clinicians, payers, manufacturers and researchers can work together to improve the quality and efficiency of clinical research to benefit decision-making in clinical and health policy. These initiatives are made possible by the active collaboration of numerous public and private sector experts, stakeholders and policymakers.
Pragmatic clinical trials
One method of addressing evidence gaps is through the expanded use of Pragmatic Clinical Trials (PCTs)—prospective controlled studies designed specifically with the objective of assisting patients, clinicians and payers in making informed decisions about alternative medical therapies. A number of important characteristics generally distinguish pragmatic trials and traditional clinical trials. First, PCTs involve the deliberate selection of clinically relevant alternative interventions for comparison, chosen based on the most common decision-making scenarios. Many trials do not include highly relevant comparison arms, leaving decision makers to depend on less reliable, indirect comparisons with which to make clinical and policy choices. Second, PCTs are designed to make the results as generalisable as possible, and therefore are more likely to include a more diverse population of study participants. While many important studies have been done with narrow inclusion and exclusion criteria, one of the common ways in which many RCTs fail to address important questions is by unnecessarily excluding patients with common co-morbidities and demographic characteristics, making the application of results to individual real-world patients more challenging. Third, PCTs select outcomes that are intended to address the primary issues and concerns of patients, clinicians and payers. Many RCTs include outcomes that are of primary interest to regulators, and pay less attention to the post-regulatory decision makers that will also use those studies to guide their choices. These outcomes may include more quality-of-life information, and may involve longer follow-up periods than are typical for traditional clinical trials. Selecting the most useful and relevant outcomes requires direct consultation with decision makers during study protocol development. In fact, one of the keys to the successful design of clinical trials that are more useful for decision making is the greater engagement of decision makers in trial design.
CMTP has been working on methods for developing pragmatic clinical trials, and has recently begun a project in collaboration with experts, stakeholders and policy makers to create a conceptual, methodological and policy framework that will improve the design and implementation of pragmatic clinical trials for phase III pharmaceutical trials. This initiative will clarify the nature of evidence desired by decision makers, explore methodological approaches to the design of phase III trials, identify regulatory, methodological, business, and other challenges to PCTs, and discuss potential strategies to overcome these challenges.
Effectiveness guidance documents
Another approach to improving the link between the evidence desired by decision makers and the output of the clinical research enterprise is to develop a shared understanding of the nature of the desired evidence. CMTP has begun to develop a library of Effectiveness Guidance Documents (EGDs), which are analogous to the guidance documents issued by the U.S. Food and Drug Administration, intended to provide product developers and clinical researchers with guidance on the design of clinical studies intended to support regulatory approval. In contrast, EGDs provide recommendations for study designs about specific categories of technologies that are intended to provide healthcare decision makers with a reasonable level of confidence that the technology will improve healthcare outcomes. Current topics under development include gene expression profiling for cancer risk prediction, cardiac imaging and treatment of chronic wounds.
The target audience for EGDs is similar to the audience for FDA guidance documents—clinical researchers and product developers. The process for developing these documents involves integrating the perspectives of the full range of stakeholders, including consumer, payers, clinicians, product developers, regulators, researchers and others. By setting clear prospective standards for evidence, decision makers can increase the chances that these recommendations will be incorporated into clinical studies, and that those studies will be more likely to produce the information that all of these stakeholders consider most relevant.
While EGDs have no legal or binding effect on any decision maker or stakeholder, their influence would derive from the transparency, creditability, neutrality and technical accuracy associated with the iterative multi-stakeholder development process. Product developers would not be required to design studies in accordance with the relevant EGD, and payers would not be bound to those principles in making coverage decisions. Nonetheless, these documents should reduce some of the uncertainty about what sort of evidence decision makers are looking for when considering the use of new technologies.
Coverage with evidence development
Decision makers in the public and private health insurance industries have long been faced with the problem of making coverage decisions for ‘promising’ but unproven medical technologies. Frequently, they are torn between the demands of patients and their physicians for innovative healthcare techniques, and the desire to have definitive evidence about the clinical and comparative effectiveness of the new technology. For most new technologies, substantial questions exist about their optimal use for many years after they are initially introduced, and the incentive for these questions to be addressed is substantially reduced once payment has been secured. In 2005, the Center for Medicare and Medicaid Services (CMS), the federal agency that provides health insurance to special populations within the US, began a programme called ‘coverage with evidence development’ (CED.) CED was a new approach to offer coverage for promising technologies under the condition that patients participate in a registry or clinical trial, which would generate clinical evidence that could be used at a future date for more definitive decision making and coverage decisions. While CED has its share of challenges to overcome as the programme is further refined, it has the potential to be an effective approach to allowing rapid coverage decisions while still generating valuable evidence for future decision making. CMTP is currently working with private payers as well as a range of other stakeholders in the US to develop a policy framework for private sector CED. The goal is to establish a routine process by which important emerging technologies can be identified for CED, and adequately designed studies can be developed. Individual health plans can then make a decision to participate in a given CED initiative, and the actual research will be sub-contacted to an independent and credible research organisation.
Conclusions
Important gaps in evidence for decision making have now become widely recognised, and this was a major factor behind the recent decision of the US Congress to provide US$ 1.1 billion to support comparative effectiveness research. In order for this money to be spent effectively, it will be important to have a meaningful and sustained collaboration between researchers and decision makers in deciding on research priorities, establishing methodological standards, developing methods that accurately reflect important questions and developing a sustainable framework to guide and support the work. CMTP has been working for the past several years to develop some specific tools and strategies to facilitate comparative effectiveness research. It is our hope that these ‘targeted interventions’ will address the ‘defective transport mechanisms’ that prevent communication between ‘intracellular’ decision makers and the ‘extracellular’ clinical research enterprise.

