

Heart failure is a major health problem worldwide. Current therapies manage the symptoms of heart failure. New experimental findings suggest possible future therapies that arrest the development of heart failure.
Heart failure afflicts 1-2 per cent of the general population and 6-10 per cent of the elderly (<65 years) population in developed nations. The five-year survival rate for heart failure patients is 50 per cent. There is a growing incidence of reported heart failure in other parts of the world including Asia, Africa and the Middle East. Pathological cardiac hypertrophy exhibits increased ventricular-wall tension and cardiac contractile dysfunction, and is a major predictor of heart failure.
The current treatments of heart failure address normalisation of its symptoms by using pharmacological agents to improve contractile performance. For example, drugs such as digitalis can improve beat-to-beat force generation, drugs such as Angiotensin Converting Enzyme (ACE) inhibitors and β-blockers modulate neurohumeral signalling to enhance contractile performance, and drugs such as calcium channel blockers normalise cardiac calcium handling. Furthermore, it has been shown that exercise helps to normalise cardiac function by reversing some of the systemic changes seen during heart failure. In spite of these therapies, there currently exist no approved drugs that can reverse or prevent the development of heart failure.
In order to understand the disease progression of heart failure, it is necessary to understand the changes to heart contractile function at the cellular level. Heart failure is characterised by changes in the expression of the proteins governing ionic currents and calcium handling. This is most easily studied in laboratory animals. Examples of pathological cardiac hypertrophy in laboratory animals include agonist-stimulated hypertrophy, hormone-induced hypertrophy (e.g. Norepinephrine (NE), Phenylephrine (PE), or Isoproterenol (ISO), and pressure-overload hypertrophy induced by banding of the thoracic aorta.
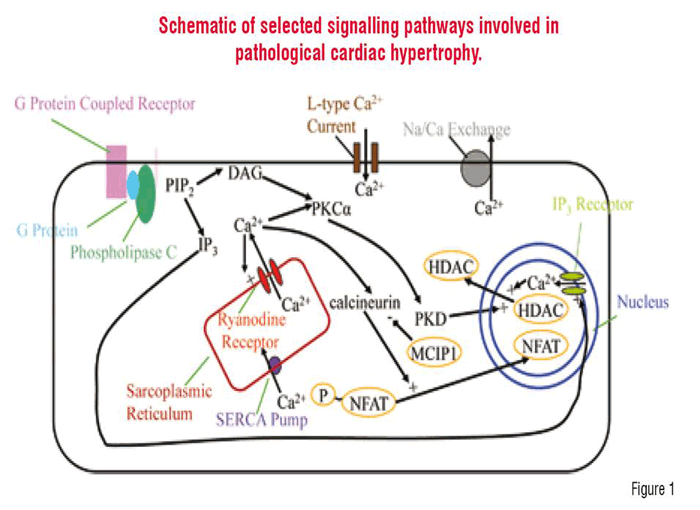
The physiological symptoms of cardiac hypertrophy and heart failure are accompanied in part by renewed expression of the fetal cardiac genes i.e. there is dysnormalisation of gene expression concomitant on the disturbed signalling that cascades in diseased cardiomyocytes. Translocation of the transcription factor NFATc2 (nuclear factor of activated T-lymphocytes) to the nucleus has been shown to be required for the induction of hypertrophy. Export of the signalling protein class II HDAC (histone deacetylase) has been observed to accompany the translocation of NFAT into the nucleus. The elevation of intracellular calcium is a common regulatory factor in both NFAT import into the nucleus and class II HDAC export from the nucleus. The activation of NFAT requires the calcium / calmodulin dependent activation of calcineurin, which dephosphorylates NFAT allowing its import into the nucleus (Figure 1). In fact, calcineurin has been implicated as a necessary signalling molecule in the induction of cardiac hypertrophy. In the heart, the cytosolic calcium concentration is elevated periodically as calcium is elevated to cause contraction of the myocyte. A number of investigators have suggested that the periodic release of contractile calcium can trigger activation of the transcription factors that lead to hypertrophy and heart failure. In fact, there are several lines of evidence that suggest this. For example, the rapid pacing (tachycardia) of the canine ventricle by an implanted electrode (about four beats a second or 4 Hz) for prolonged periods has been used to induce hypertrophy and heart failure. Furthermore, other work has shown that in neonatal rat ventricular myocytes, exposure to angiotensin II caused an increase in the rate of contractile cytosolic calcium transients and an increase in the amount of NFAT translocated to the nucleus. Additionally, these investigators also showed that increasing the pacing of neonatal ventricular myocytes by field stimulation also resulted in an increase in NFAT translocation to the nucleus. Activation of NFAT leads to the expression of MCIP1 that acts to inhibit calcineurin. Experiments have shown that overexpression of MCIP1 inhibits cardiac hypertrophy. Furthermore, NFAT transclocation to the nucleus can be blocked by immunosuppresants such a cyclosporin A and FK506. Unfortunately the use of immunosuppresants would leave the patient immunocompromised.
Another important class of transcription factor involved in the advent of heart failure are proteins that regulate the histone proteins. The DNA in the nucleus is tightly wrapped around histone proteins. Histone Acetyltransferases (HATs) acetylate the histones allowing portions of the DNA to become accessible for transcription. Histone Deacetylases (HDACs) oppose the action of HATs. Nuclear class I HDAC inhibits anti-hypertrophic gene transcription. Inhibitors of class I HDAC such as tricostatin A, sodium butyrate, and HC-toxin block cardiac hypertrophy and improve contractility in failing hearts. Nuclear class II HDAC inhibits pro-hypertrophic gene transcription, in fact, nuclear class II HDAC is thought to block the activation of NFAT-activated genes. Thus, it has been proposed that normalising cardiac gene expression with small molecules, such as class II HDAC inhibitors, will halt cardiac remodelling processes by controlling the disrupted signalling cascades, and thus might be a successful ‘transcriptional therapy’ for the failing heart. In fact, knockout of class II HDACS produced spontaneous hypertrophy in mice and antiviral overexpression of active mutants of class II HDAC prevented agonist-induced hypertrophy in cultured myocytes.
The export of class II HDAC requires activation of calmodulin kinase II and local calcium release from perinuclear IP3. Hypertrophic growth can be blocked by expression of a calmodulin kinase II resistant mutant HDAC in COS cells. Nuclear export of HDAC-GFP in isolated myocytes in response to endothelin-1 activation can be blocked by the IP3 receptor antagonist 2-APB. In these studies, contractile calcium transients were not sufficient to cause HDAC export from the nucleus suggesting another source of calcium, such as release of calcium from the nuclear envelope triggered by inositol 1,4,5-trisphosphate (IP3).
Protein kinase D also regulates HDAC through a related pathway as follows: Activation of phospholipase C, typically in response to agonist cleaves phosphatidyl inositol into Diacylglycerol (DAG) and IP3. DAG activates protein kinase C which activates protein kinase D which in turn promotes class II HDAC export from the nucleus. DAG kinase phopshorylates DAG and hence decreases its concentration. This reduction in DAG concentration reduces class II HDAC export from the nucleus through protein kinase C and D. Additionally, DAG kinase has been shown to suppress cardiac hypertrophy and loss of left ventricular function in mice. It blocks endothelin-1 induced growth and activation of fetal gene programs caused through the PKC pathway.
Inhibition of HAT, which opposes the action of HDAC, leads to reduced DNA transcription of hypertrophic proteins. Curcumin is a natural polyphenolic compound found in turmeric that does just that. Curcumin blocks phenylephrine- and pressure overload-induced cardiac hypertrophy in primary cultured rat cardiac myocytes through inhibition of P-300 HAT activity. Hence, this provides another potential mechanism to control hypertrophy.
There are also other pathways that might be exploited. For example, resveratrol is a non-flavinoid polyphenol found in the skin of red grapes. Resveratrol activates the AMP-Activated Protein Kinase (AMPK) and inhibits Akt pathways. Resveratrol inhibits pressure overload- or phenylephrine-induced heart failure in rats. This drug also has other potential applications. For example, resveratrol is currently under phase II clinical trials for prevention of colon cancer.
In summary, recent experiments have given insight into the complex signalling pathways associated with cardiac hypertrophy and heart failure. These suggest potential drug targets and lead compounds that warrant further study. Due to the complexities of the signalling pathways, further studies are likely to additional drug targets and lead compounds.










