





Requirements for the regulation of development, propagation and approval of medical devices are of great competitive interest, which assures that it should be of good quality, safety and efficacy, in order to protect, improve and monitor public health. After the development and before the distribution of medical devices into the market, it is supposed to be licensed by respective regulatory authorities across the globe. Since the 1980s, it has been reported that there was an abrupt change in the worldwide regulation of medical devices, as the manufacturers were forced to fulfil the regulatory requirements, documented standards, norms, guidelines, specifications, testing methods for the design, and manufacturing of devices. As a consequence, the figure has shifted from very few to 60-65 countries for the implementation of the regulation. The Global Harmonization Task Force (GHTF) came into existence in 1993, aiming to systematise the medical device regulation across the sphere. It was replaced by International Medical Device Regulatory Forum(IMDRF) in 2011 which was concerned with the future aspects of ‘Harmonized medical device regulation’ which involves number of regulatory authorities of Australia, Brazil, Canada, China, Europe, Japan, Russia, Singapore, South Korea, and US under the supervision of World Health Organization (WHO), 1948.
Medical Devices
According to the Federal Drugs and Cosmetics Act, ‘a device is an instrument, apparatus, implement, machine, contrivance, implant or an in-vitro reagent’ that should encounter three conditions: 1)It should acknowledge the official National Formulary or the US Pharmacopoeia. 2) It should be deliberately used in the diagnosis, cure, alleviation, treatment, and prevention of disease. 3) Lastly, it is intended to be used to alter the structure and functionality of human body. Hence use of medical devices has a number of important roles including: 1) It plays a crucial role in improving, protecting, supporting or sustaining life 2) It assists in diagnosis, prevention, monitoring, cures and mitigates disease or injury, 3) It is widely used in research, substitution, alteration and even in the support of the anatomy and physiology of the human body, 4) It is also employed for control of conception 5) last but not the least, it provides knowledge related to medical aspects by means of in vitro testing of specimens obtained from human body. It has been estimated that an average of 3-7 years is spent for the approval of medical devices; drugs, on the other hand, take approximately 12 years.
Need for Medical Device Harmonisation
Medical device harmonisation aims to ‘encourage convergence in regulatory practices related to ensuring the safety, effectiveness, performance and quality of medical devices, promoting technological innovation and facilitating international trade’. Harmonising medical device is essential because it decreases the time span in marketing of products, cuts the cost required to market a product, enhances the efficacy of government as it facilitates cooperation among regulators and even in organising activities of regulation, improves trading and extent of marketing and raise up the public health protection. Hence this leads to the increment of safety and efficacy of device, thus, it lifts up the public health and also gains consumer’s faith and confidence. A comparative study of the five regions in aspect of regulation of medical devices is in Table1.
United States
In the US, medical devices come under the cloud of United States of Federal Drug and Administration(USFDA). If a device is sanctioned by the FDA, it reflects to be safe and effective for the intended purpose, it can thus be legally introduced into the market. It uses the ‘least burdensome approach’, which means that the manufacturers are supposed to present the only that data which are essential to show the device’s safety and efficacy. The devices are classified into three categories on the basis of level of risk as follows:
i. Class I - low risk devices, e.g., gauze and tooth brushes.
ii. Class II - moderate risk devices, e.g., suture and needles.
iii. Class III- significant risk devices, e.g., pacemakers, implantable defibrillators.
For acceptance of devices in US three regulatory pathways are followed:
1) Pre-market notification(PMN) pathway: PMN is also known as 510(k) clearance process and it comes under the section of medical device amendments of 1976. This pathway is for the regulation of those devices which are classified under class I and some (25 per cent) under class II. It is a submission shown to the FDA for the demonstration of the device’s safety, efficacy and substantially equivalence to predicate device (device which is already available in the market). According to Code of Federal Regulation (Title 21, Section 807), if a device is to be cleared through this pathway then the manufacturer is supposed to present that the device is substantially equivalent to the predicate device. Hence, it does not require any clinical data, so it is less time consuming and less expensive than Pre-Market Approval(PMA). Due to this reason it is also called as fast track approval process. Substantial equivalence does not refer to the similar identity of a new and a predicate device. In fact it means that a new device’s ‘intended use’ and “technological characteristics” should be same to that of the predicate device. The intended use means the primary function of device which further includes its “indication of use” which explains that the device should diagnose or treat the disease or condition which it is intended to. And for the estimation of technological characteristics the manufacturer is supposed to submit a description of design, material of construction, and other technological features of device. In July 2011, the Institute of Medicine (IoM) reported that the process is only concerned with the substantial equivalence to an existing device and is least bothered about its safety and efficacy. Due to this drawback they recommended the abolishment of the process, which forced the manufacturers to provide prior clinical data to support the clearance but the data is not parallel to that of PMA.
Comparative study of the five regions in aspect of regulation of medical devices
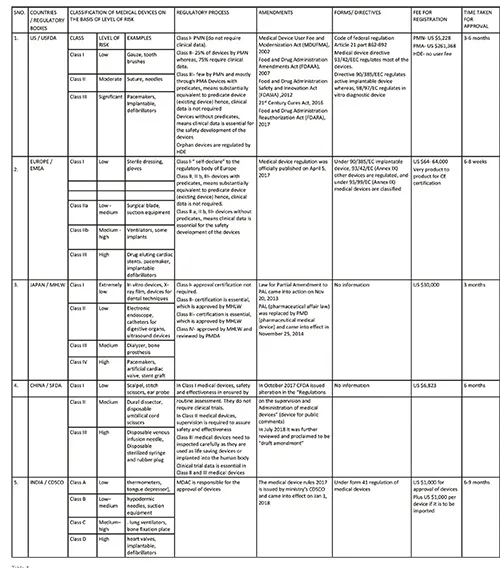
2) Pre-market Approval (PMA) pathway: For the evaluation of a device’s safety and effectiveness, the PMA pathway is followed. It is responsible for the regulation of most (75 per cent) of the class II and the entire for class III devices. It is only applicable to those devices which are non-identical to predicate devices. Thus, such devices are termed as ‘De Novo Devices’. This involves clinical trials that further provide data essential for manufacturing, approval and adoption of devices. It requires sufficient clinical data for the approval, therefore, it is the strictest device marketing application than PMN. The PMA submission must include information related to use, manufacturing, reference, and performance standards or voluntary standards, outcomes of all non-clinical and clinical studies. This pathway involves four different stages which contribute to the regulation of medical devices, they are: i. Pre–Investigational Device Exemption/Pre-clinical evaluation, ii. Clinical testing, iii. PMA submission, iv. Post marketing surveillance. These stages are summarised in figure 1.

Comparision between PMN [510(k)], PMA and HDE
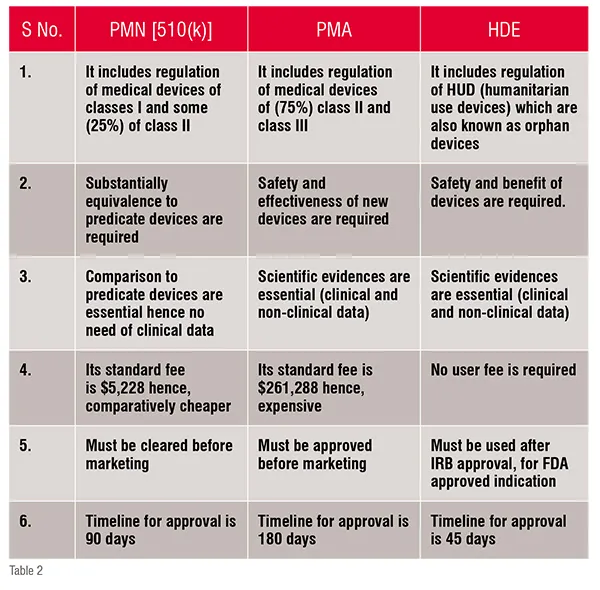
3) Humanitarian Device Exemption (HDE) pathway: Devices that are used to treat or diagnose rare disorders (fewer than 4000 individuals per year) are included in HDE. The Safe Medical Device Act 1990 and its rules were amended by FDA in 1996, which motivated the manufacturers to produce devices used in rare disorders. It requires to show safety but not the efficacy of the device. The content and form of this application is similar to that of PMA application except in context of effectiveness requirement. The office of orphan product development regulates HDE through FDA. Besides FDA, IRB (Institutional Review Board) supervises Humanitarian Use Devices (HUD) regulation and approval.
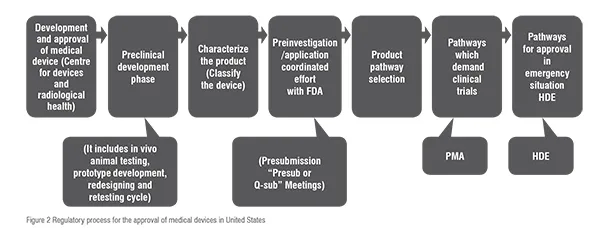
The three pathways involved for the approval of medical devices in US are summarised and compared in Table2. Thereby for the approval of devices firstly the classification of device is identify and according the belonging class, the regulatory pathway is followed in order verify whether the device is suitable to place into market and hence the device is approved under USFDA. The sets of procedures are briefly summarised in figure: 2.
Europe
In Europe, medical devices approval follows the path of ‘harmonisation’ along with the guidelines of the European Union (EU). Medical device regulatory procedures rely on Medical Device Directives (MDD), which in turn consist of three core directives, which are amended for safe regulation and marketing of medical devices. They are: 1) implantable devices are regulated under directive 90/385/EC (European Commission); 2) most of the other devices are regulated under directive 93/42/ EC; 3) in-vitro diagnostic devices are regulated under 98/99/EC. EU 2017/745 is the new regulation which in freshly published; in the coming 3 to 5 years, these three directives will be replaced by this regulation. As per Directive 93/42/EC, a medical device is defined as “any instrument, apparatus, appliance, material or other article, whether used alone or in combination, including the software necessary for its proper application intended by the manufacturer to be used for human beings for the purpose of diagnosis, prevention, monitoring, treatment or alleviation of disease; diagnosis, monitoring, treatment, alleviation of or compensation for an injury or handicap; investigation, replacement or modification of the anatomy or of a physiological process; control of conception; and which does not achieve its principal intended action in or on the human body by pharmacological, immunological, or metabolic means, but which may be assisted in its function by such means”. Annex IX of the MDD 93/40/EEC categorises medical devices into four classes on the basis of the level of risk according to the patient condition and requirement. They are as follows:
i. Class I: low risk devices, e.g., sterile dressing and gloves.
ii. Class IIa: low-medium risk devices, e.g., surgical blades, suction equipment and radiotherapy equipment.
iii. Class IIb: medium-high risk devices, e.g., ventilators and some implant.
iv. Class III: high risk devices, e.g., drug eluting cardiac stents, pacemakers and implantable defibrillators.
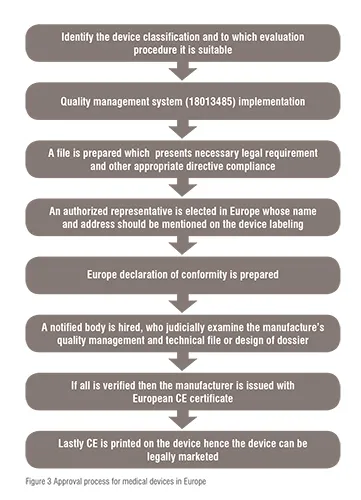
Class I (low risk) devices are ‘self-declared’ or ‘self marked’ which means that the manufacturer itself approves the compliance and applies CE (Conformité Européenne) mark and thus can be placed into European market (according to Annex VII Module A, EC Declaration of Conformity). In case of some devices of Class IIb and Class III (device with predicates) there is no requirement of clinical data. Class IIa, IIb, and III (devices without predicates) require clinical data to ensure safety. Some class III devices (high risk) require clinical trial data to demonstrate the safety and performance of the device. Furthermore, the devices (except class I devices) are approved through a ‘Decentralised approval process’. In this process the application can be filed in any member state and is further reviewed by Notified Bodies (NBs). These bodies were set up within the state and were authorised by a competent authority or health agency.
In Europe there are in total 50 NBs, these NBs are private organisations which sign the agreement with the manufacturers and charge a fee for the certification of the devices. The company is free to choose any NB for the review of a particular class of device. The NB inspects carefully the application and makes sure that it fulfils the EC regulation. If the regulatory necessities are met by the devices then it issues the CE mark to the devices. Hence, medical devices marked CE can be sold in any country in Europe. This mark clearly indicates that the product is compliant to relevant legislation of safety. This action by the NB is termed as “conformity assessment” in Europe. The marketed device under the sanction of CE mark does not require any further evaluation but, the new regulation of 2010 has made the requirement for approval of devices tighten in aspect of similarity of new device to that of redicate device (device already in the market), and proactive post marketing surveillance. The Decentralised Approval process is summarised in figure 3.
Japan
Regulation of medical devices in Japan is a combination of the regulatory processes of US and Europe. Pharmaceutical Medical Device Act (PMDA) is responsible for the review and approval of the process. Medical devices are classified into four classes based on the level of risk as follows:
i. Class I: extremely low risk devices, e.g., in-vitro devices, x-ray films and devices for dental techniques
ii. Class II: low risk devices, e.g., electronic endoscopes, cathetersfor digestive organs and ultrasound devices
iii. Class III: medium risk devices, e.g., dialyser and bone prosthesis
iv. Class IV: high risk devices, e.g., pacemakers, artificial cardiac valves and stent graft.
Devices belonging to class I are of extremely low risk and requires a submission of marketing called ‘Todokede’ which means certification for approval is not required. On the other hand, class II devices of low risk and some medium risk devices of class III should have compliance directly related to the norms and standards required for the approval certification. ‘Ninsho’ is the application to be submitted for the approval of device which will be evaluated by registered certification, which is further recommended by the Ministry of Health, Labor and Welfare(MHLW). Rest of the devices of class II and IV undergo prior review by the PMDA, and further approval is given on the basis of the MHLW proposal. The submission applications for such devices are called ‘Shonin’ which is quite similar to that of a PMA submission in USA. The devices are manufactured under the disciplines of quality systems compliant with their own rules and regulation in MHLW ordinance 169. If there is no availability of office space for the companies in Japan, then there is need of an in-country representative to be signatory to the submission and a Marketing Authorization Holder (MAH) license.
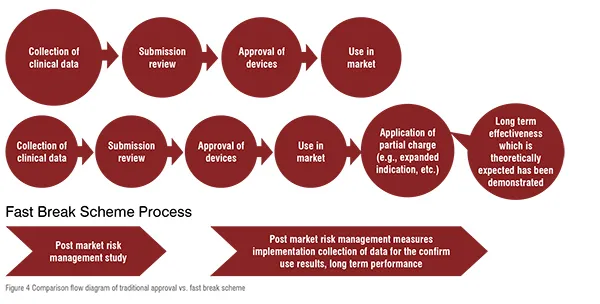
On 31 July, 2017, the MHLW formulated a new regulatory framework called fast, a break scheme for innovative medical device to accelerate patient access. The scheme is a collection of organised things required for the approval of innovative medical devices in Japan. Hence, the device should accurately progress at the initial stage of development on the basis of clinical evidence and not restricted to intense prospective randomised controlled trials, and should include clinical data which is agreeable for the prediction of clinical advantages and safety, on the basis of finite patient population in a particular clinical setting. The scheme is only applicable to ‘brand new medical devices’ which should fulfil the following requirements: 1) There should not be an already suitable alternative device unless there is a satisfactory probability of greater safety, efficacy and performance than the existing one. 2) The disease for which it is to be used should be a life threatening disease or a serious disability. 3) Some clinical evidence should be present for support. 4) Commitment of postmarketing to a suitable management and rigorous real world evidence collection and estimation. 5) If a new prospective clinical trial is not conducted, a justification should be given. The traditional approval and fast break scheme is compared in figure 2, which clearly indicates that the traditional one requires more clinical data, whereas in the fast break scheme the innovative medical devices should fulfil specified criteria or standards on the basis of existing clinical data. Thus it needs lesser data collection and ultimately consumes less time than the traditional process. The comparison between Traditional process and fast break scheme is illustrated by figure 4.
China
In China, for a medical device to be sold in the market, it must be registered with the State Food and Drug Administration (SFDA), which is presently known as China Food and Drug Administration(CFDA). There are two important regulations which should be followed in China: ‘Regulations for the Supervision and Administration of Medical Device’ (2000) and ‘Measures for the Administration of the Medical Device Registration’ (2004). The registration requirements are alike to that of the US and Europe. According to classification of devices on the basis of level of risk the medical devices are divided into three classes as follows:
i. Class I: low risk devices e.g.,scalpels, stitch scissors and ear probes
ii. Class II: moderate risk devices e.g., dural dissectors and disposable umbilical cords
iii. Class III: high risk devices, e.g., disposable sterilised syringes and rubber plugs, and disposable venous infusion needles.

Class I devices’ safety and effectiveness is assured by routine administration. Class II medical devices require further supervision for the evaluation of their safety and effectiveness. Class III medical devices need to be precisely controlled in terms of safety and effectiveness as they are used as life saving devices that are implanted into the human body. For registration of devices of Class II and Class III it is essential to present clinical trial data. The devices belonging to Class III and other imported devices are directly supervised by the CFDA. Some of the devices also need the China Compulsory Certification (CCC) apart from medical device registration. The CCC mark is handled by the Administration of Quality, Supervision, Inspection and Quarantine, a Chinese quality and quarantine authority (AQSIQ).
As per the provision of medical device registration, the application for the purpose of registration of devices can only be lifted by a legal authority of China. The manufacturer is supposed to submit a declaration that the devices withstand the Chinese National Standard and Professional/Sectorial standards without any kind of alteration, and if any, the manufacturer can add the particular requirement to the CFDA on standard directly connected to the device. The legal and technical documents are submitted to CFDA; further the product sample testing is done. Clinical trial data are collected and presented to the CFDA for Class II and III devices. The Chinese Medical Doctor Association (CMDA) conducts specialised assessment, and the final inspection is done by the CFDA for registration certification. The summary of the process of approval of medical device in China is expressed in figure 5.

India
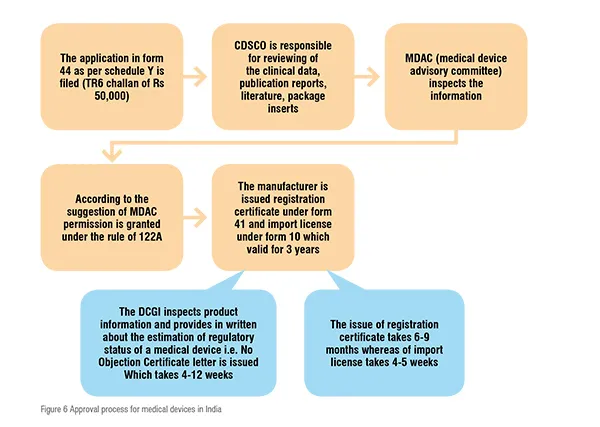
The Central Drug Standard Control Organization (CDSCO) under the Directorate General of Health Services in Ministry of Health & Family Welfare (MoHFW), Government of India(GoI), is the National Regulatory Authority (NRA) which is accountable for manufacturing, import, conduct of clinical trials, laying down standards, sale and distribution of medical devices via enforcement of medical device rules, 2017. It reviews all data submitted which are essential for the approval of the device. The NRA is supposed to ensure the safety of public health. As the NRA, the CDSCO is responsible for conducting Materiovigilance programme of India (MvPI). The Indian Pharmacopoeia Commission acts as the NCC (National Coordination Centre) for MvPI. The MvPI is supposed to furnish the collection of safety data in a systemic manner, therefore regulatory selections and guidelines for secure use of medical devices being used in India might be based totally on the data generated. The programme supervises MDAE (Medical Devices-associated Adverse Events), spread awareness among healthcare professionals regarding the significance of MDAE reporting in India, and also overlooks the advantages and negative outcomes of the medical devices. It is also intended to generate unbiased evidence-based suggestions and to communicate the findings to all key stakeholders.
The MDAC (Medical Device Advisory Committee) is a body responsible for the overall inspection of the information. If everything is in favourable the MDAC grants permission under the rule of 122A and ultimately the manufacturer is issued with license under form 41 and the import license under form 10 allowing them to introduce the device into the market. The license issued is valid for only 3 years. The devices which are non-notified need not to be registered with the CDSCO and can be imported throughout the country according to the formal custom rules. The sets of procedure of device regulation are summarised in figure 6.
Currently in India regulation on import of medical are imported from Europe and the US because of their better quality and efficiency. The import and registration of medical devices occurs under Drug and Cosmetics Rules, 1940 which specify that i) In order to file an application for the import and registration of medical devices under the prescribed guidelines, the importer is given a time period of 60 days. ii) Import is not allowed without the approval of an authorised body, in case of those devices that has never imported in the country before the date of notification. iii) The devices are allowed to be sold for a particular period (up to 6 months) during which the application is being evaluated for approval or rejection. Separate committees are made for the assessment of different classes of devices. The distinct medical device classes categorised on the basis of level of risk are:
i. Class A- Low risk devices e.g., thermometers, tongue depressor
ii. Class B- Low–medium risk devices e.g., hypodermic needles, suction equipment
iii. Class C- Medium–high risk devices e.g., lung ventilators, bone fixation plates
iv. Class D- High risk devices e.g. heart valves, implantable defibrillators.
Conclusion
Regulation for the approval of medical devices across the globe is a vital requirement in order to ensure quality, safety, efficacy and performance so that they can be introduced into the market for protecting, preventing, improving, and sustaining public health. This ultimately lifts up the customer’s faith and confidence in the device and in the manufacturer. The devices are classified on the basis of the level of risk into various classes, thus the devices in different classes have different regulatory steps for approval of the devices varying from region to region. Although different regions are diverse in the regulatory processes, charge different fee for application filing, and have their own time period in order to approve and to issue a license, at the end all are aiming at one goal, that is manufacturing and marketing of a device which is safe, effective, and of good quality in order to provide their best to the public.
REFERENCES:
1. Gail A. Van Norman. Drugs and Devices Comparison of European and U.S. Approval Processes. Basic to transitional science. 2016; 1(5): 245-302X, 399-410.
2. Kaushik A, Saini KS1, Anil B, and Rambabu S. Harmonized Medical Device Regulation: Need, Challenges, and Risks of not harmonizing the Regulation in Asia. J Young Pharm. 2010; 2(1): 10.4103/0975-1483.62221, 101-106.
3. Carmelo De Maria, Licia Di Pietro, Andrés DíazLantada, June Madete, Philippa Ngaju Makobore, Mannan Mridha, Alice Ravizza g, Janno Toroph, Arti Ahluwalia. Safe innovation: On medical device legislation in Europe and Africa. Health Policy and Technology. 2018; 7(2): 156–165.
4. Gail A. Van Norman, MD. Drugs, Devices, and the FDA: Part 2 An Overview of Approval Processes: FDA Approval of Medical Devices. Basic to transitional science. 2016; 1(4): 452 - 302 X, 277-287.
5. Radhadevi N, Balamuralidhara V, Pramod Kumar TM, Ravi V. Regulatory guidelines for medical devices in India: An overview. Asian J Pharm. 2012; 6: 10.4103/0973-8398.100125, 10-17.
6. Ashley Adamovich, MD, Susie Park, MD, Gary P. Siskin, MD, Meridith J. Englander, MD, Kenneth D. Mandato, MD, Allen Herr, MD, and Lawrence J. Keating, MD. The ABCs of the FDA: A Primer on the Role of the United States Food and Drug Administration in Medical Device Approvals and IR Research. J VascInterv Radiol. 2015; 26189046, 1-7.
7. Frances J. Richmond, Sai S. Tatavarty. Medical device and diagnostic products. 2018, 105-136.
8. Jonathan P. Jarow, John H. Baxley: Medical devices: US medical device regulation. Urologic Oncology: Seminars and Original Investigations. 2014; 25458071, 1-5.
9. Akihide Konishi, SoichiroIsobe, Daisaku Sato, Akihide Konishi. New Regulatory Framework for Medical Devices in Japan. Current Regulatory Considerations Regarding Clinical Studies. J Vasc Interv Radiol. 2018; 29548874, 1-4.
10. Shunsuke Matsushita, Keisuke Tachibana and Masuo Kondoh. The Clinical Innovation Network: a policy for promoting development of drugs and medical devices in Japan. Drug Discovery Today. 2018; 10.1016, 1-4.
11. Ganesh GNK, Nishanthini. B, Lakshmi T. Vidhya. Medical Device Regulation in US, Europe, China and India. International Journal of Drug Regulatory Affair. 2017; 5(2): 2321-6794, 17-25.
12. Kashyap UN, Gupta V, Raghunandan HV. Comparison of drug approval process in United States and Europe. J Pharm Sci Res. 2013; 5(6): 0975-1459, 131–136.
13. Naghshineh N, Brown S, Cederna P, et al. Demystifying the U.S. Food and Drug Administration: understanding regulatory pathways. Plastic Reconstr Surg. 2014; 134(3), 559–569.
14, Fargen KM, Frei D, Fiorella D, et al. The FDA approval process for medical devices; an inherently flawed system, or valuable pathway for innovation? J Neurointerv Surg 2013; 5: 269–75.
15. Quesada O, Yang E, Redberg RF. Availability and dissemination of results from US Food and Drug Administration-mandated post approval studies for medical devices. JAMA Intern Med. 2016; 176: 1221–23.
16. Nidhi B. Agarwal and ManojKarwa. Pharmaceutical Regulations in India.2018, 215-231.
17. Naishi K, Dilip M. Documentation requirements for generic drug application to be marketed in India- a review. JPSBR 2014; 4(4): 237-42.
18. Sweet BV, Schwemm AK, Parsons DM. Review of the Processes for FDA Over sight of Drugs, Medical Devices, and Combination Products. JMCP 2011; 17: 40-50.
19. Browning P. Differences between medical devices and drugs. Br J Nurs 2014; 23(S12): S28–9.
20. Nadimpalli Radhadevi, Veeranna Balamuralidhara, Teggina Math Pramod Kumar, Valluru Ravi. Regulatory guidelines for medical devices in India: An overview. Asian Journal of Pharmaceutics 2012; 6(1):1998-409x, 10-17.
21. Kramer DB, Xu S, Kesselheim AS. Regulation of medical devices in the United States and European Union. N Engl J Med. 2012; 366: 848–55.
22. Maresova P, Penhaker M, Selamat A, Kuca K. The potential of medical device industry in technological and economical context. Ther Clin Risk Manag 2015; 11: 1505–14.
23. Cyranoski D. Japan to offer fast-track approval path for stem cell therapies. Nat Med 2013; 19(5): 510.
24. Kondo H, Hata T, Ito K, et al. The current status of Sakigake designation in Japan, Prime in the European Union, and Breakthrough Therapy Designation in the United States. Ther Innov Regul Sci 2017; 51:1–4.
25. https://www.slideshare.net/mobilesamNixonS/medicaldevice-regulation-71076618 (Accessed on October 15, 2018).
26. http://www.slideshare.net/mobile/chetanUmale/medical-device-registration-in-india-37039083 (Accessed on October 16 2018).
27.https://www.tuvseed.com/industries/medical-devices-healthcare/market-approval-amp-certification/eu-mark (Accessed on October 18 2018).
28. https://www.asiaactual.com/japan/medical-device-registration/ (Accessed on October 18 2018).


